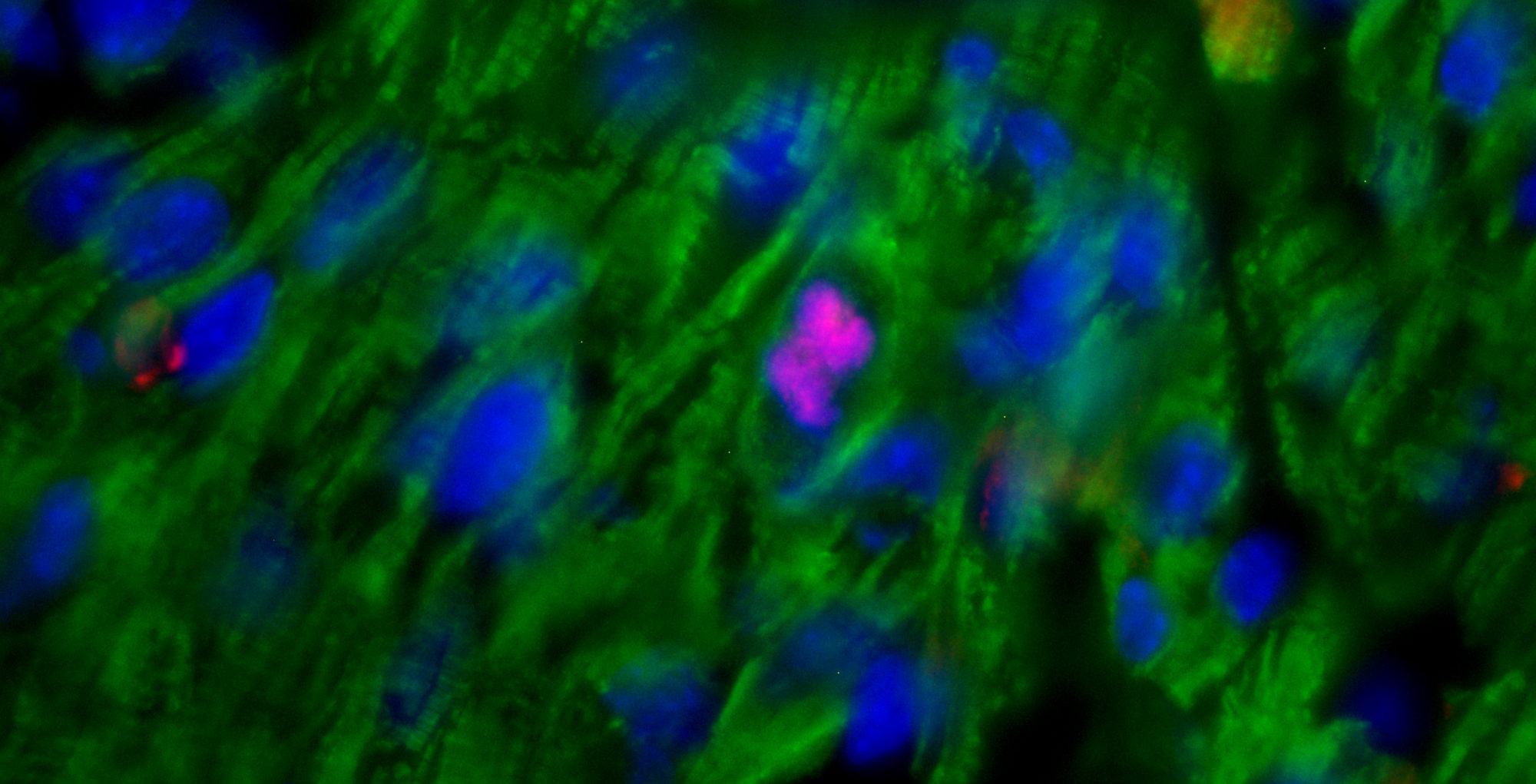
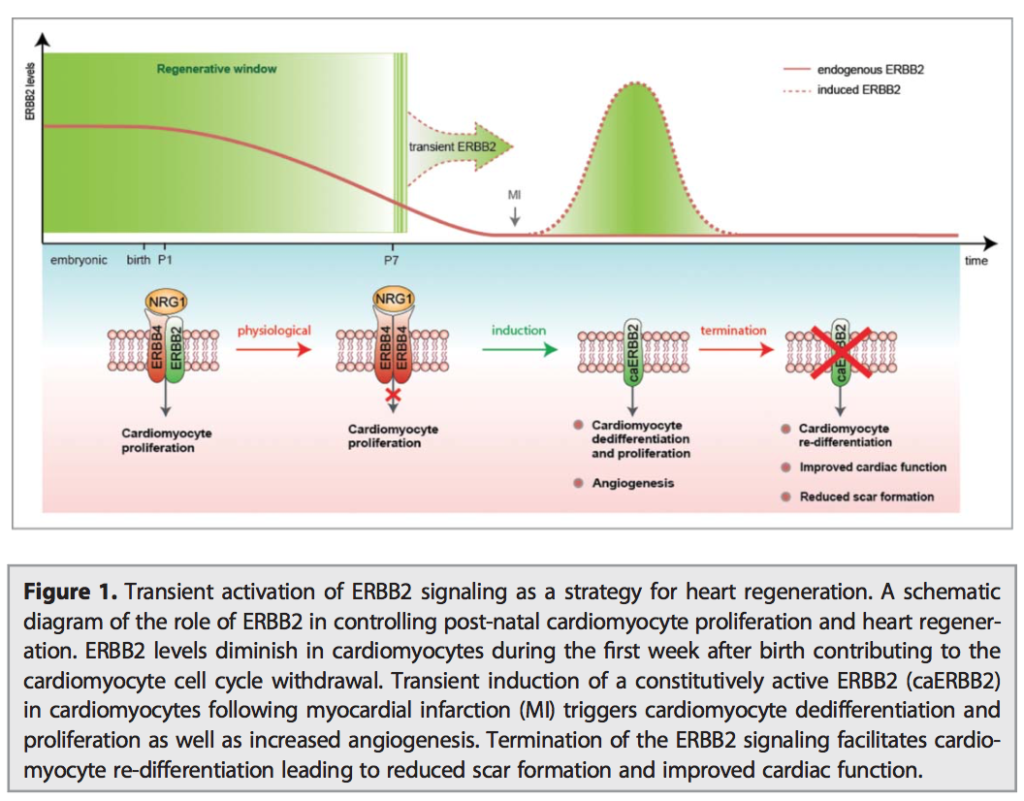
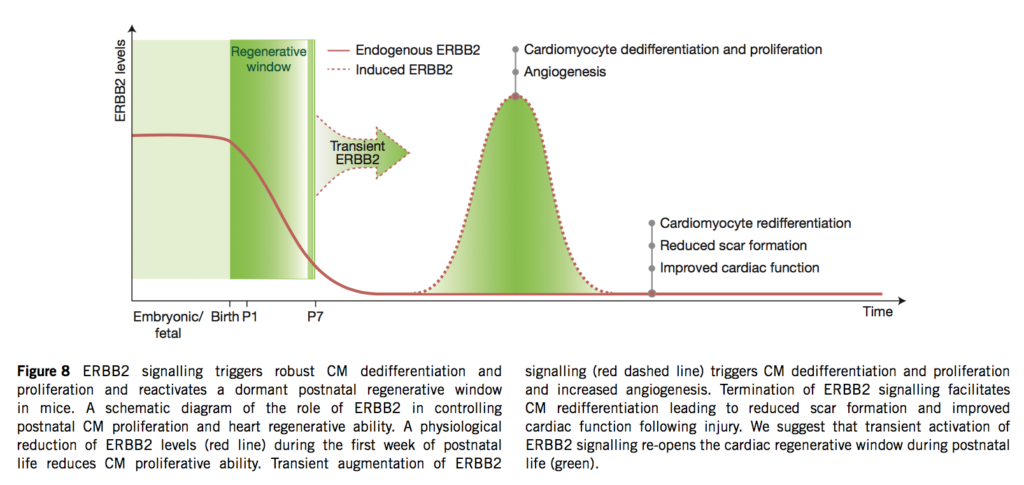
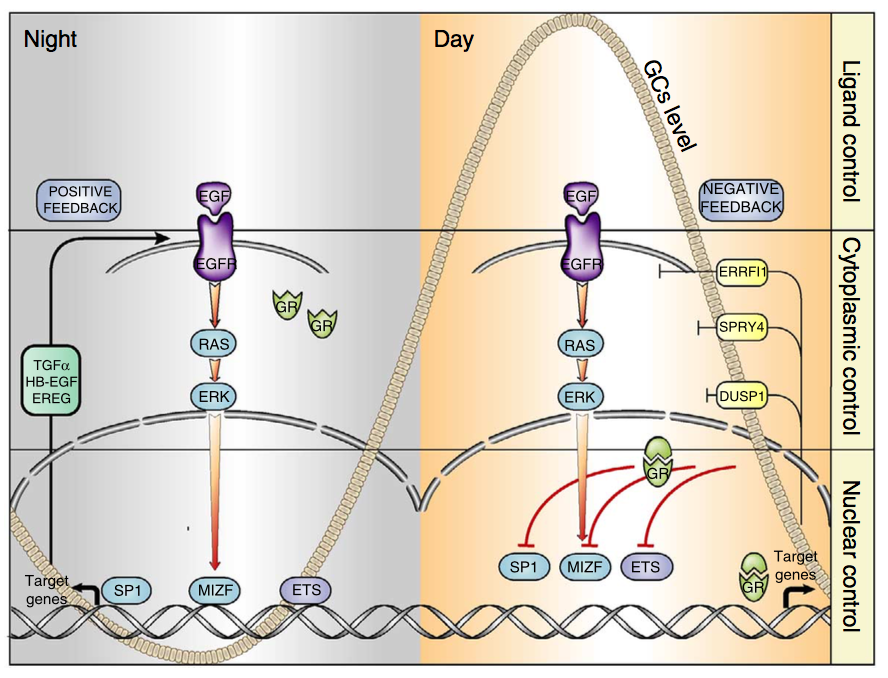
06.04.2015 – The Guardian: Heart muscle cells regrown in medical research breakthrough
06.04.2015 – The Sydney Morning Herald: Australian researchers help find way to regrow heart muscle
07.04.2015 – RT: Scientists discover revolutionary method to regrow heart muscles
07.04.2015 – AM: Helping the heart repair itself after a cardiac arrest: researchers say they’ve worked out how
07.04.2015 – Financial Express: The Turbo-charging hormone can repair ‘broken’ heart
07.04.2015 – El Comercio: Logran regenerar músculos del corazón al estimular una hormona
07.04.2015 – SKY NEWS: Breakthrough Sees Heart Muscle Cells Regrown
07.04.2015 – SBS: Scientific breakthrough could give new hope to heart-attack patients
07.04.2015 – The Jewish Press: Israeli-Australian Researchers Discover How to Regrow Heart Muscle
07.04.2015 – Business Magazin: STUDIU: Cercetătorii au reuşit să stimuleze regenerarea celulelor muşchiului cardiac
07.04.2015 – iarul de Iasi: Cercetătorii au reuşit să stimuleze regenerarea celulelor muşchiului cardiac
07.04.2015 – ZeeNews: Scientists regenerate cardiac muscles through hormone stimulation
07.04.2015 – Medical Xpress: Research effort leads to mammalian heart tissue regeneration
07.04.2015 – Kurier: Durchbruch: Forscher lassen Herzmuskelzellen wachsen
08.04.2015 – SBS World NEWS: Researchers Trigger Heart Regeneration After Heart Attack (TV news)
08.04.2015 – The Times of Israel: Medical leap as Israeli researchers regrow heart cells
08.04.2015 – smarTherapy: Consiguen regenerar los músculos del corazón mediante estimulación hormonal
08.04.2015 – Scimondo: Aktivierung eines Hormon-Signalweges hilft Mäuseherzen nach einem Herzinfarkt auf die Sprünge
08.04.2015 – Beyond the Dish: Dead Heart Muscle Regrown in Rodents
08.04.2015 – Phys.org: Research finds turbo-charging hormone can regrow the heart
08.04.2015 – Newsmax: Hormone Found to Regenerate Heart Muscle
09.04.2015 – Medical Daily: Heart Attack Patients May Regrow Cardiac Cells By 2020 Thanks To Breakthrough Discovery
09.04.2015 – The Australian: Bashing corporate Australia has reunited the faceless man with his Green frenemies
10.04.2015 – SILICONWADI: Ricercatori israeliani fanno ricrescere le cellule del cuore
10.04.2015 – Italiasalute.it: Nuova tecnica per curare il cuore infartuato
10.04.2015 – Tiscali: Cuore infartuato, scoperto il meccanismo che avvia la formazione di nuove cellule
10.04.2015 – Ansa: Scoperta la chiave per riparare il cuore infartuato
10.04.2015 – MeteoWeb.eu: Salute: scoperta la chiave per riparare il cuore infartuato
10.04.2015 – Bionn: 拯救心脏 拒绝梗死! – 大健康产业专区- 生物谷
11.04.2015 – Giornale di Sicilia: Cuore colpito da infarto, scoperta la chiave per ripararlo
12.04.2015 – Il Quotidiano di Puglia: Con questo gene riparerò il cuore
12.04.2015 – NurseTimes: Scoperta la chiave per riparare il tessuto cardiaco infartuato
13.04.2015 – Il Sole 24 Ore: Cuore, identificato gene capace di “ripararlo”
13.04.2015 – HealthCanal: Heart Cells Regenerated in Mice
13.04.2015 – IBA: הלב את מחדשים
13.04.2015 – Radio Jai: Científicos de Israel regeneran células del corazón de ratones
13.04.2015 – EnlaceJudío: Científicos israelíes regeneran células cardíacas
13.04.2015 – israel21c: Weizmann Institute scientists regenerate heart cells in mice
13.04.2015 – IsraCast: Medical Breakthrough – Israeli Scientists Find Way to Regrow Heart Muscle Cells
13.04.2015 – Science Daily: Heart cells regenerated in mice
13.04.2015 – Popular Science: Scoperta la chiave per riparare il cuore infartuato
13.04.2015 – Science Codex: Heart cells regenerated in mice in NGR1 study
13.04.2015 – Science 2.0: Heart Cells Regenerated By Going Backward To Make Progress
13.04.2015 – ScienceBlogs – The Weizmann Wave: Guest post: Dr. Gabriele D’Uva: How to Grow New Heart Cells
13.04.2015 – Il Resto del Carlino: Nuove cellule grazie alla ricerca. Così si ripara il cuore infartuato
14.04.2015 – The Jerusalem Post: Weizmann Institute researchers regenerate heart cells in mice
14.04.2015 – Farmacia.it: Lotta all’infarto: il cuore potrà essere riparato
14.04.2015 – ANI News: Regeneration of heart cells possible in mice
14.04.2015 – Business Standard: Regeneration of heart cells possible in mice
14.04.2015 – New Kerala: Regeneration of heart cells possible in mice
14.04.2015 – Corriere del mezzogiorno: Studio il cuore, sogno l’Italia
14.04.2015 – Israel Hayom: Israeli scientists make revolutionary discovery, regenerate heart cells
14.04.2015 – Genetic Engeneering & Biotechnology News: Old Heart Cells Divide Like New
14.04.2015 – Daily News & Analysis: Scientists regenerate heart cells in mice
14.04.2015 – Cardiovascular Disease News: Study Advances Understanding of Heart Cells’ Regeneration
14.04.2015 – Comité Central Israelita Uruguay: Científicos israelíes regeneran células cardíacas
14.04.2015 – Prahova: Cercetatorii au reusit sa stimuleze regenerarea celulelor muschiului cardiac
14.04.2015 – Iran Daily: Heart cells regenerated in mice
14.04.2015 – Horizon 2020 projects: ERC-funded research sees mouse heart cells renewed
14.04.2015 – Israeli Ministry of Foreign Affairs: Heart cells regenerated in mice
14.04.2015 – MedIndia: Regeneration of Heart Cells Possible in Mice With the Help of Specialized Protein, ERBB2
14.04.2015 – DAILYROUNDS: Successful heart muscle regeneration in mice may soon be seen in humans
14.04.2015 – Jns: Israeli Scientists Regenerate Heart Cells in Revolutionary Discovery
15.04.2015 – The Algemeiner: Israeli Scientists Regenerate Heart Cells in Revolutionary Discovery
15.04.2015 – Nature子刊:ERBB2触发哺乳动物心脏再生是通过促进心肌细胞去分化和增殖
15.04.2015 – Sanità Salento: Un cuore rigenerato per gli infartuati (con intervista radiofonica)
15.04.2015 – Medical Insider: Ученые научились восстанавливать сердечную мышцу
16.04.2015 – Diario del web: Scienziati rigenerano le cellule del cuore
17.04.2015 – Innovations Report: Weizmann Institute Scientists Regenerate Heart Cells in Mice
18.04.2015 – Globus Magazine: RIGENERAZIONE CARDIACA: TROVATA LA CHIAVE PER RIPARARE IL CUORE INFARTUATO
19.04.2015 – Ultime Tecno-scientifiche: COSI’ SI RIPARA IL CUORE INFARTUATO
19.04.2015 – Corriere di Bologna: Gabriele, il ricercatore che ha riacceso il cuore e vuol tornare in Italia
20.04.2015 – Giannella channel: È italiano il medico che ha scoperto come riparare un cuore infranto
20.04.2015 – PolskieRadio: Komórki serca potrafią się zregenerować po zawale
21.04.2015 – multibriefs: Researchers regenerate heart cells in mice
23.04.2015 – UNIBO MAGAZINES: Ecco il gene chiave per riparare il cuore dopo un infarto
23.04.2015 – UNI news24: ERBB2: scoperto il gene che rigenera il cuore dopo un infarto
23.04.2015 – La Repubblica: Trovato un gene per riparare il cuore dopo un infarto
23.04.2015 – RaiNews: Un gene ripara-cuore
23.04.2015 – Rai3 – TGR Emilia Romagna (TV news – min. 15:34)
23.04.2015 – Bologna2000.it: Identificato un gene chiave capace di riparare il cuore danneggiato da un infarto
23.04.2015 – SassuoloOnline: Identificato un gene chiave capace di riparare il cuore danneggiato da un infarto
23.04.2015 – L’Adige: Scoperto gene per riparare il cuore dopo l’infarto
23.04.2015 – Controcampus.it: Gene chiave salva il cuore dopo un infarto, ricerca Unibo
23.04.2015 – Virgilio Notizie: Scoperto un gene per riparare il cuore dopo un infarto
23.04.2015 – TRC: Post infarto, importante scoperta medica
23.04.2015 – AGI: Ricerca Unibo: identificato gene per riparare cuore dopo infarto
23.04.2015 – BolognaToday: Ricerca Unibo: scoperto gene chiave per riparare il cuore dopo un infarto
27.04.2015 – Viversani: Scoperto un gene per riparare il cuore dopo un infarto
27.04.2015 – ResearchItaly: Identified the gene that can repair the heart after a heart attack
27.04.2015 – Biotechin: A “Hearty” discovery – Turbo-charging hormone can regrow the heart
28.04.2015 – Salzburger Nachrichten: Krebs-Wachstumsrezeptor steuert Regeneration von Herzzellen
29.04.2015 – HealthCanal: HEART ATTACK BREAKTHROUGH
02.05.2015 – LiveUniversity: Scoperto gene chiave capace di riparare il cuore danneggiato da un infarto
06.05.2015 – The Australian Jewish News: A heartening collaboration
06.05.2015 – Iton Gadol: Researchers Regenerate Heart Cells In What Could Be A Huge Breakthrough For Heart Disease Treatments
06.05.2015 – AJN: Avances. Investigadores israelíes regeneran células del corazón para tratar la enfermedad cardiac
06.05.2015 – NoCamels: Researchers Regenerate Heart Cells In What Could Be A Huge Breakthrough For Heart Disease Treatments
06.05.2015 – vetscite.org: Heart cells regenerated in mice
07.05.2015 – Yad be Yad: Investigadores israelíes regeneran células del corazón para tratar enfermedades cardíacas
08.05.2015 – Classic Chaos: Researchers Regenerate Heart Cells In What Could Be A Huge Breakthrough For Heart Disease Treatments
14.05.2015 – VitAssistance: Cuore, identificato gene capace di ripararlo
6.06.2015 – Ruthfully yours: Amazing Israel :Researchers Regenerate Heart Cells In What Could Be A Huge Breakthrough For Heart Disease Treatments